
AI and high-throughput data-enabled gene circuit design
Rai et al. demonstrate the first-time use of AI and high-throughput sequencing for genetic circuit design in mammalian cells.

Just over two weeks ago, Kshitij Rai and Ronan O’Connell at Rice University published a paper introducing CLASSIC, a genetic screening platform that can profile over 100,000 gene circuit candidates in a single experiment. Using data generated by CLASSIC, the authors train machine learning models that can predict the function of a genetic circuit based on the nucleic acid ‘parts’ it is composed of. CLASSIC, and the data-driven genetic circuit design it enables, demonstrate that ML and high-throughput screening can be used to design genetic circuits with desired behaviors more affordably and quickly, a crucial step toward building economically viable SynBio products.
The authors demonstrate CLASSIC (“combining long- and short-range sequencing to investigate genetic complexity;” what an acronym!) on the problem of designing high-fold change circuits: DNA constructs that allow cells to produce large amounts of a fluorescent protein in the presence of a specific chemical (inducer), and have a low expression level (basal expression) without the inducer present.
Instead of designing and testing a few different options, the authors build a massive set of candidate constructs by combinatorially assembling genetic ‘parts’ (promoters, coding sequences, terminators, etc.).1 Using a relatively small number of parts, they form a circuit design space of 165,888 possible genetic constructs.
Each of these constructs (also called libraries) includes an identifying barcode sequence. Barcodes are linked to specific libraries using long-read sequencing (i.e. sequencing the entire construct, which contains the library and the barcode). Then, each of the libraries are integrated into an individual cell (in this case, human embryonic kidney (HEK293T) cells). The cells are sorted based on how well they adhere to the design specification, then short-read sequencing2 is used to identify the barcode sequences in each of the cells, and thereby the libraries they contain. In this way, circuit composition can be linked to phenotype at a massive scale.
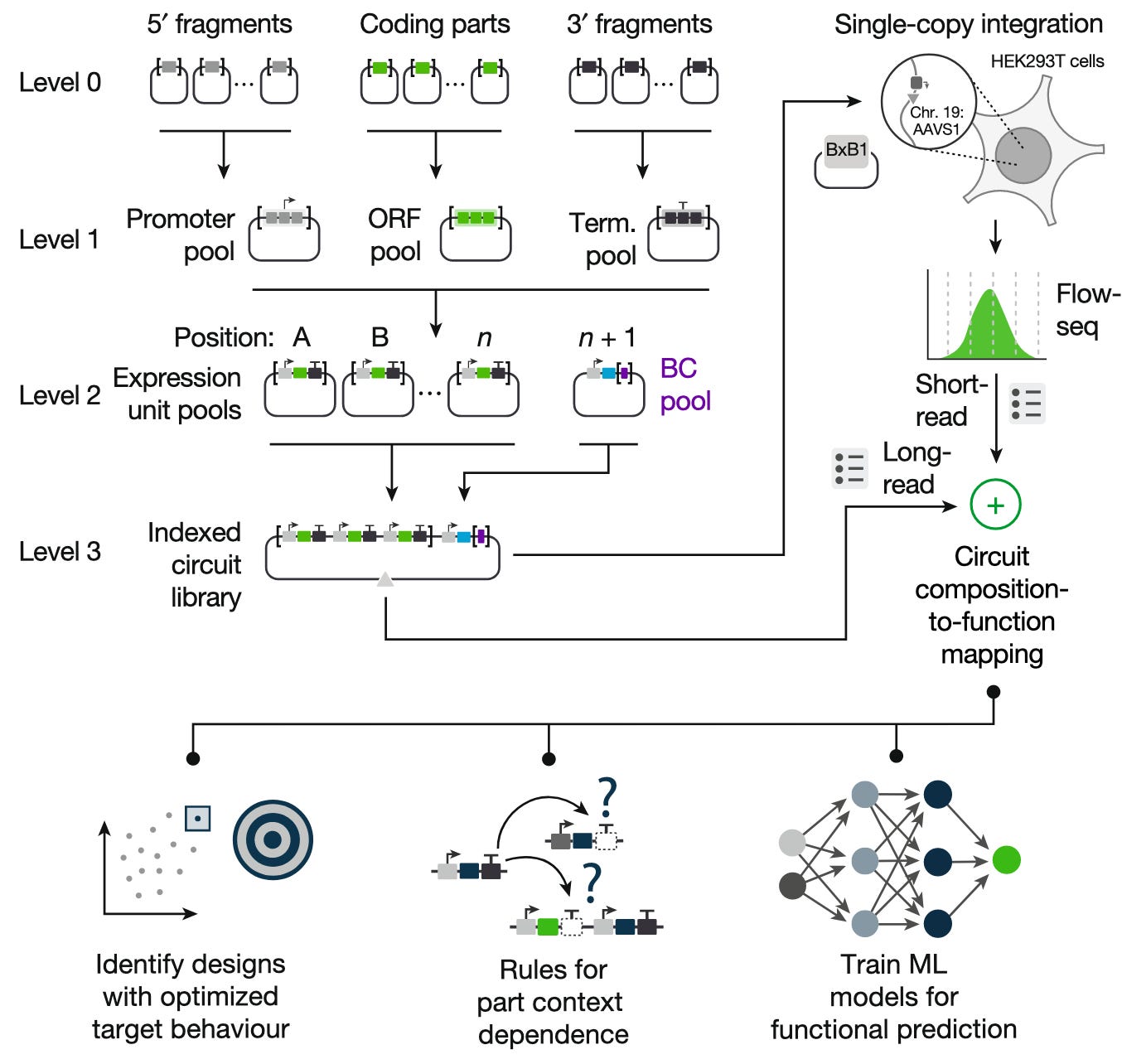
CLASSIC workflow. (Figure 1b, Rai et al.)
Of the 165,888 possible circuits that could have been generated given the genetic part options, however, only 73% were successfully tested and profiled. To predict the function of all other possible circuits (~40,000) in this particular circuit design space, Rai et al. train a multilayer perceptron on the data obtained through CLASSIC and experimentally validate the constructs that the model predicted would have the highest fold change in expression (60-80x). Notably, they find that their MLP “significantly out-perform[s]” a biophysically relevant model (a fact that they use to claim that more advanced (machine learning) models are required to capture complex, nonlinear biological interactions).
Data-driven methods, the authors propose, also promise to help discover rules for genetic circuit design. How to derive rational design rules for genetic circuits is an open problem in synthetic biology. Genetic circuits are highly context-dependent (what is the host organism? what genes and biomolecules are being used? in what environment is the engineered cell deployed?) making it extremely difficult to write down a small set of rules that can be applied universally to design new gene circuits.
“CLASSIC-enabled design space mapping indicates that … [high fold change] does not arise from a single optimal solution, but from multiple balanced combinations of parts that achieve similar phenotypic outcomes through subtly different molecular strategies, with medium-activity components providing the most reliable overall route to circuit performance … This demonstrates the utility of using a data-driven approach to solve complex circuit design challenges and identify designs that support target circuit behaviour, while revealing underlying, non-intuitive design principles that are challenging to capture using biophysical models.”
summary + thoughts on AI vs. biophysical models
The benefits of CLASSIC (and the data-driven learning it enables) are twofold:
At a foundational level: CLASSIC generates high-quality data that can be used to infer patterns or rules about how to design circuits in a particular context.
From an applications standpoint, this high-throughput approach allows for testing hundreds of thousands of circuit variants at once, greatly reducing the number of iterative cycles required to build a genetic circuit that meets certain specifications. This feels very similar to small-molecule drug discovery (where millions of candidate molecules are screened until there’s a hit).
Rai et al. perform several other experiments using CLASSIC and demonstrate the use of machine learning for other applications beyond composition-to-function prediction, but this first example is enough to convey the benefits of their combined approach of high-throughput experimentation with machine learning methods.
Does this mean that biophysical modeling will be left in the dust? (I don’t think so.) While the algorithms used here identify correlations between circuit composition and function, they do not offer a mechanistic understanding of why those compositions are optimal. Perhaps we can combine biophysical and data-driven methods to develop a coarsely interpretable model (but this is for a future post!).
Find the paper here:
Rai, K., O’Connell, R. W., Piepergerdes, T. C., Wang, Y., Brown, L. B. C., Samra, K. D., Wilson, J. A., Lin, S., Zhang, T. H., Ramos, E. M., Sun, A., Kille, B., Curry, K. D., Rocks, J. W., Treangen, T. J., Mehta, P., & Bashor, C. J. (2026). Ultra-high-throughput mapping of genetic design space. Nature, 1–10. https://doi.org/10.1038/s41586-025-09933-9
At a basic level, a gene needs a promoter (a region of DNA where molecular machinery binds to initiate transcription into RNA), a coding region (the DNA that codes for the protein product), and a terminator (a sequence that signals to the end of transcription to the cellular transcription machinery). Together, these components form an expression unit (this sequence of DNA is capable of allowing a cell to express a protein). Not all promoters and terminators are made equal; for example, transcription enzymes may bind more strongly to certain promoter sequences over others. By testing different combinations of these three genetic parts, the authors seek to build a genetic circuit that best fits their specifications. The authors study a small gene regulation network consisting of three genes that regulate each other; each construct that is introduced into a human cell has three expression units.
Short-read sequencing is a cheaper and faster alternative to long-read sequencing that produces reads that are about 50-300 bases long. By contrast, long-read sequencing produces 1,000-10,000 base reads. The authors use long reads to map each barcode to its library's full sequence, and short-read sequencing is used to cheaply read out barcodes.
The sheer scale of 100,000 gene circuit candidates profiled by CLASSIC really stood out. This builds so powerfully on the innovative, data-driven thinking I've come to appreciate from your writing. Using ML to predict circuit function from nucleic acid 'parts' is truly elegant and such a game-changer for SynBio cost-efficiency. It's a brilliant application of ML to biological design, making complex systems much more accesible.